Abbreviations
- LVH: Left ventricular Hypertrophy
- HCM: Hypertrophic Cardiomyopathy
- SAM: Systolic Anterior Motion of Mitral Valve
- LV: Left ventricle
Background
- Hypertrophic Cardiomyopathy is a genetic cardiomyopathy (prevalence approximately 1:500) caused by mutations in the sarcomere genes and has variable phenotypic expression
- Hallmark of HCM is left ventricular hypertrophy that is not explained by an alternate cause.
- NOTE: The definition of HCM based on AHA 2020 is:
“A disease state in which morphologic expression is confined solely to the heart. It is characterized predominantly by LVH in the absence of another cardiac, systemic, or metabolic disease capable of producing the magnitude of hypertrophy evident in a given patient and for which a disease-causing sarcomere (or sarcomere-related) variant is identified, or genetic etiology remains unresolved.” – AHA 2020 HCM Guideline
- Management of HCM consists of:
- Treating symptoms of reduced cardiac output
- Risk of sudden cardiac death
Diagnosis
- Generally diagnosed by transthoracic echocardiography (MRI if inadequate image quality)
- Definition
AHA 2020 HCM Guideline Diagnosis of HCM - Absence of alternate cause of LVH
AND - Ventricular wall thickness of ≥15 mm
OR
- Ventricular wall thickness of ≥13 mm if family history or confirmed gene mutation of HCM
- Absence of alternate cause of LVH
- NOTE: There are many causes for “thick walls” on cardiac imaging, which can be due to primary genetic (HCM), secondary due metabolic condition, or physiologic due to changes in hemodynamics.
- Secondary causes must be ruled out clinically before HCM is diagnosed. Most of them are ruled out with imaging and clinical history.
- For example, if the patient has severe aortic stenosis, the diagnosis of HCM is uncertain and unlikely.
- Diagnosis can be challenging in athletes, elderly patients with isolated basal septal hypertrophy and those with family history but also alternate pathology (ie. hypertension).
Differential Diagnosis
- Primary: Hypertrophic Cardiomyopathy
- Secondary:
- Amyloidosis (TTR, AL)
- Metabolic Disorders
- Lysosomal Storage Disease (Anderson-Fabry, Hurlers)
- Glycogen Storage Disease (Forbes, Danon)
- Carnitine Deficiency, PKB, Mitochondria Myopathy
- NOTE: Red flags: movement disorder, peripheral neuropathy, and renal dysfunction
- Syndromic
- i.e. Noonan’s Syndrome, LEOPARD Syndrome, Friedreich’s Ataxia
- Physiologic:
(physiologic adaptation due to change in LV loading parameters – usually somewhat reversible)- Aortic stenosis
- Hypertensive heart disease (usually mild LVH)
- Athlete’s Heart
- Other findings that are common but not diagnostic are:
- systolic anterior motion of the mitral valve
- hypertrophied and apically displaced papillary muscle
- myocardial crypts
- elongated mitral valve leaflets
- myocardial bridging
- right ventricular hypertrophy
Types
- Hypertrophy distribution
- Distribution of LVH can be in any pattern but is classically asymmetric with disproportionate involvement of the septum. However concentric (symmetric) hypertrophy variants are described.
- Most common presentation is LVH at the septum but also can present as concentric hypertrophy or isolated apical hypertrophy
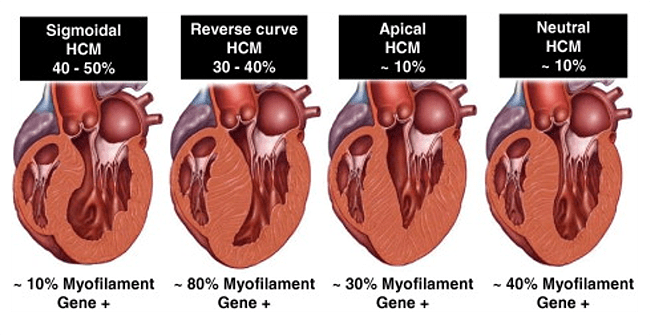
Obstructive vs. Non-Obstructive
- Hypertrophic Obstructive Cardiomyopathy (HOCM) – refers to a subtype of HCM with clinically significant obstruction of the LV outflow tract (LVOT).
- Patients with obstructive HCM have a higher risk of sudden death, are generally more symptomatic, and obstruction has important clinical management implications.
Genetics
- Genetic testing is used to identify at risk family members and to screen for HCM mimickers such as Fabry’s disease
- Most common genes with mutations for HCM are MYH7 (beta-myosin heavy chain 7) and MYBPC3 (myosin binding protein-C3) which account for 75% of the patients though 50% of patients do not have an identifiable gene
- Other genes include TNNT2, TNNI, TPM1, ACTC and others that are still being discovered
- Mutations in sarcomere are autosomal-dominant thus first-degree family members have a 50% risk of carrying the mutation
- Those with identified genetic sarcomere mutation have worse outcomes (Study)
- Note: LVH rate of development varies per patient (even within families) and for some may never occur
Family Counselling
- First-degree relatives should be offered clinical screening (see below) and genetic testing (when pathologic mutation found)
Presentation
- Patients generally present with:
- Symptoms of reduced cardiac output (see pathophysiology): shortness of breath, reduced exercise tolerance, exertional syncope/presyncope.
- Symptoms of arrhythmia: Syncope, presyncope, palpitations
- Symptoms of volume overload (end-stage)
- Incidentally discovered on cardiac imaging.
Pathophysiology
- LV Outflow Tract (LVOT) obstruction
- Occurs due to thick septal muscle, which obstructs outflow from the LV, and mitral valve abnormalities (longer leaflets and anterior displaced papillary muscles)
- A harsh outflow murmur on physical exam is classic for Obstructive HCM.
- LVOT obstruction is DYNAMIC, meaning it can vary substantially based on preload/afterload conditions:
- Any maneuver that increases LV volume/pressure opens the LVOT, and reduces obstruction. This REDUCES the intensity of the murmur. Examples are:
- Increase Preload: Stand to squat, Passive leg raise, Hydration, early valsalva
- Increase Afterload: Isometric hand grip, inflated bilateral BP cuffs.
- Any maneuver that reduces LV volume/pressure reduces the diameter of the LVOT, which increases obstruction. This produces a larger pressure gradient across the LVOT, and the murmur intensity is INCREASED.
- Reduce Preload: Squat to stand, dehydration, medications (i.e. nitroglycerin), mid/late valsalva
- Reduce Afterload: Medications (i.e. vasodilators)
- Any maneuver that increases LV volume/pressure opens the LVOT, and reduces obstruction. This REDUCES the intensity of the murmur. Examples are:
- NOTE: LVOT Obstruction is DYNAMIC, which is different than obstruction in aortic stenosis (AS). The murmur in obstructive HCM has much larger variability in intensity because its very dependent on LV loading parameters. In fact it can even be absent some days. Also, an AS murmur can be expected to increase in intensity or remain unchanged with maneuvers that increase preload, which is opposite to the response in a patient with obstructive HCM.
- LVOT Obstruction is measured on echocardiography by measuring the pressure gradient in the outflow tract (velocity of blood flow):
- Peak LVOT gradient of ≥30 mm Hg is considered the cutoff for obstruction.
- Mitral regurgitation
- Caused by Systolic Anterior Motion (SAM) or primary leaflet abnormalities
- MR related to SAM is often mid-late systole and posterior directed
- Diastolic dysfunction
- Myocardial ischemia
- Causes include microvascular disease, high intracavitary pressure (hyperdynamic systolic function + LVOTO), atherosclerosis and myocardial bridging
- Arrhythmias
- Atrial fibrillation and ventricular arrhythmias
- Autonomic dysfunction
- Impaired heart rate recovery and inappropriate vasodilation
- Systolic dysfunction (i.e. reduced EF)
- Uncommon but can occur late in disease (aka “burnt out HCM”) (study)
Clinical Evaluation
- Comprehensive medical/cardiac history
- Key symptoms: chest pain, shortness of breath, palpitations, and syncope
- Physical exam (including maneuvers such as Valsalva, squat-to-stand, passive leg raising, or walking)
- Systolic murmurs (holosystolic murmur over mitral area from MR and/or crescendo-decrescendo murmur over LLSB from LVOTO)
- Prominent point of maximal impulse (may be bifid)
- S4
- Abnormal carotid pulse (pulsus bisferiens or two peaks)
- 3-generation family history
- ECG
- Cardiac Testing
- Echo every 1-2 years
- Holter every 1-2 years (identify arrhythmias and SCD risk)
- MRI at initial assessment and every 3-5 years (diagnosis if inconclusive echo or possible alternate causes and SCD risk)
- Stressing testing every 2-3 years or if symptomatic
- Genetic testing

Echocardiography
See topic Echo in Hypertrophic Cardiomyopathy (pending)
Management
Medical Management
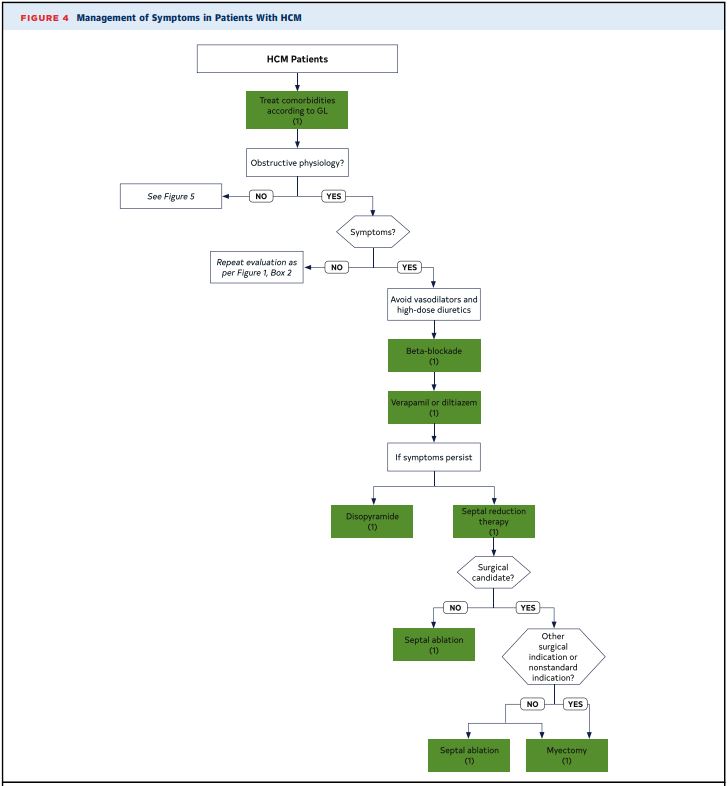
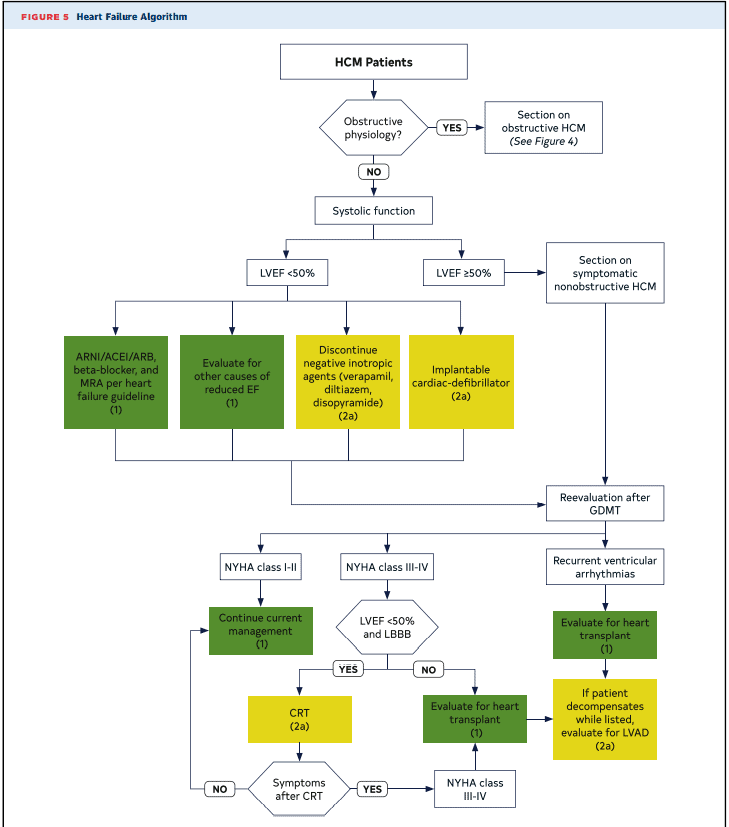
- First step is to consider mechanism and cause of symptoms (ie. chest pain from HCM vs co-existing coronary disease)
- No medication shown to alter natural history (Asymptomatic patients should be managed by close clinical observation)
- LVOT Obstruction
- Non-pharmacologic:
- Avoid volume depletion, tachycardia and vasodilatory drugs (these drugs lower intracavitary volume/pressure, and improve LVOT obstruction)
- Pharmacologic:
- 1st Line: Beta blockers
or 2nd Line: Non-dihydropyridine Calcium channel blockers (diltiazem/verapamil) - NOTE: Negative inotropes such as beta-blockers and CCBs reduce contractility of the septal muscle, which reduces LVOT obstruction. Positive inotropes do the opposite.
- Refractory symptoms:
- Disopyramide, which is a Class 1A anti-arrhythmic (monitor QTC) (works as a negative inotrope)
- 1st Line: Beta blockers
- Invasive (Surgical):
- Surgical intervention is only for patients who are symptomatic despite maximal medical therapy.
- Option 1 (preferred): Septal reduction surgery
- Option 2 (if poor surgery candidate): Alcohol septal ablation
- Decision between surgery and alcohol septal ablation should be made with the surgeon.
- Non-pharmacologic:
- Chest Pain or Shortness of Breath
- Beta blockers or Calcium channel blockers
- Heart Failure
- Diuretics for congestion
- If EF < 50% treat as reduced EF as per guidelines and investigate for alternate cause (ie. CAD)
- Atrial Fibrillation
- Anticoagulation recommended with DOAC regardless of CHADS score for clinical AF or >24 hours of subclinical AF
Invasive Management of LVOT Obstruction
- Septal reduction therapy (myectomy or alcohol septal ablation) indications for LVOTO (Peak gradient ≥50 mm Hg at rest or provoked)
- Class I:
- Severe shortness of breath or chest pain (NYHA III-IV )
- Exertional syncope/presyncope
- Symptomatic and undergoing other cardiac surgery
- Class II:
- NYHA II if other clinical factors ie. severe/progressive PH, LAA with symptomatic AF, poor functional status confirmed on treadmill
- Class I:
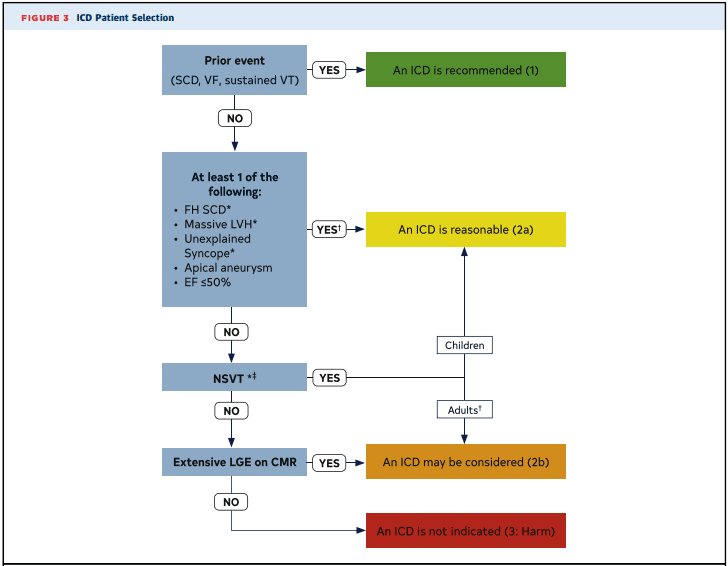
Implantable Cardioverter Defibrillator
- ICD is recommended (Class 1)
- ICD Prior event (Cardiac Arrest, VF)
- Documented Sustained VT
- ICD is reasonable (Class IIa)
- Family History of SCD
- Massive LVH (30mm)
- Unexplained worrisome syncope (not vagal attacks)
- Apical aneurysm
- EF < 50%
- ICD can be considered (Class IIB)
- Non-sustained VT
- Late gadolinium enhancement (scar) > 15-20% of myocardium on MRI
- AHA Risk Calculator (Link)
- ESC Guidelines recommend use of Risk Score Calculator (HCM RISK-SCD (link)
- Age, LVH, LA size, LVOT gradient, FamHx, NSVT, Syncope
5 year risk of SCD ICD Recommendation Notes < 4 % (Low) Not Recommended Actual risk is 1.4% in some validation studies 4-6% (Int.) Consider ICD > 6% (High) ICD Recommended
Lifestyle
- Sports:
- Mild-moderate intensity recreational exercise is recommended to maintain fitness
- Competitive sports carries increased risk of SCD and guided by shared decision making with expert provider.
- Pregnancy:
- Generally safe in clinically stable HCM but should involve shared decision making about maternal and fetal risks and maternal-fetal medicine specialist
Authors
- Primary Author: Dr. Atul Jaidka (MD, FRCPC, Cardiology Fellow), Dr. Pavel Antiperovitch (MD, FRCPC, Cardiologist)
- Reviewer:
- Copy Editor:
- Last Updated: Dec. 29, 2021
- Comments or questions please email feedback@cardioguide.ca